Projects
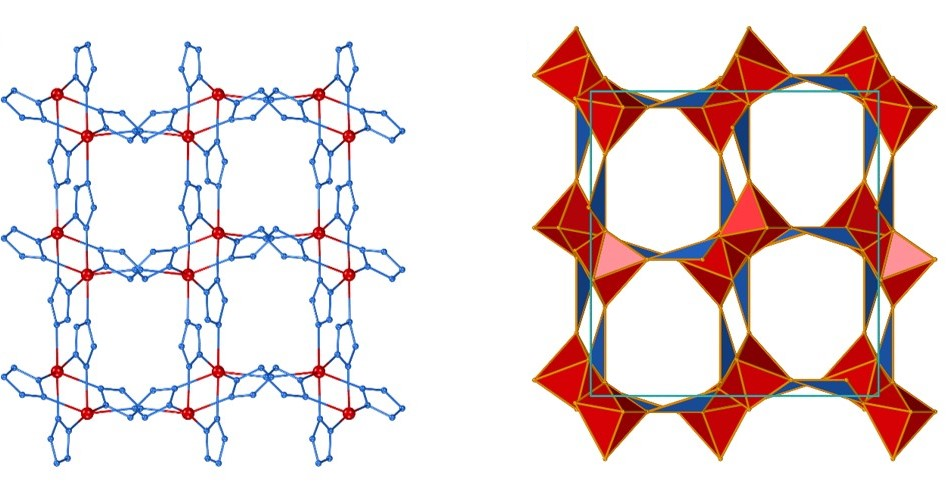
Structure prediction and polymorphism of MOFs
Understanding thermodynamic stability of metal-organic frameworks (MOFs) is crucial for designing new materials and optimizing their synthesis. DFT simulations help us explain why certain MOF topologies can be readily obtained, while others remain elusive.
The success of our predictions relies on the accuracy of our theoretical models (DFT functionals and dispersion corrections). Through a collaboration with Professor Alexandra Navrotsky (UC Davis), we have benchmarked the accuracy of DFT energies against experimental calorimetric measurements of MOF stability.
So far, we have explored the stability of zeolitic imidazolate framework (ZIF) polymorphs as a function of porosity and structure disorder, and explained the sequence of topological transformations occurring under milling conditions. Moving towards prediction of new MOF-type structures we have studied the energy landscapes of pentazolate frameworks, which resulted in the generation of several previously unreported topologies, waiting to be synthesized!
Our ultimate aim is ab initio structure prediction of MOFs, a computational algorithm capable of predicting MOF structures and their energies, starting directly from molecular diagrams of constituent nodes and linkers. In a collaboration with Dr. James Darby (University of Cambridge) and Prof. Andrew J. Morris (University of Birmingham) we have reported the first truly ab initio structure prediction of MOFs, which does not rely on any prior experimental knowledge of structure or network topology of resulting MOFs. Finally, we have demonstrated how MOF CSP can be used to design new functional MOFs, by reporting the new family of hypergolic MOFs, for which structures were first found with CSP and subsequently synthesized in a laboratory. In the future such calculations will change the way how we make MOFs, shifting from experimental screening towards theory-driven computational design.
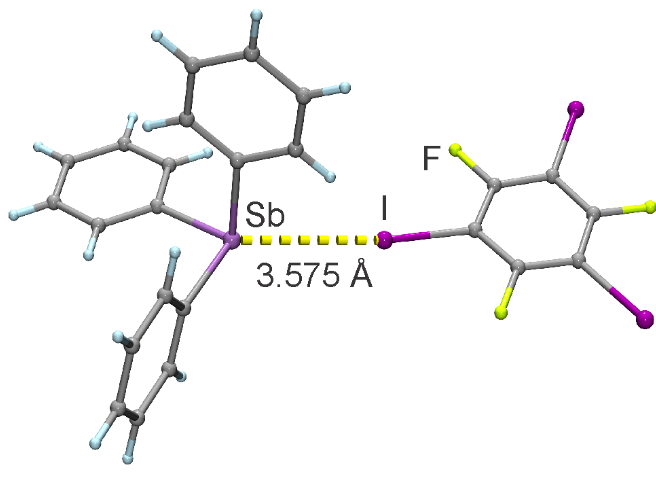
Organic cocrystals and halogen bonding
Supramolecular cocrystallization is an exciting method of bringing multiple molecules in close proximity by placing them within together within the same crystal lattice. Often this results in materials with new desirable properties, such as improved solubility, better thermal stability, efficient compressibility etc.
What is the fundamental driving force for cocrystal formation? With the aid of periodic DFT we can look at thermodynamic stability of cocrystals against their individual components, and predict the thermodynamic possibility of cocrystal interconversions. Moreover, we can explore the role of individual supramolecular interactions on the overall stability of the crystal structures.
Halogen bonding, an interaction between a heavy halogen atom electrophile and a nucleophilic atom or group, is gaining traction as a new path to construct cocrystals. Computational modelling of these interactions remains a challenge, and a choice of optimal DFT methodology remain a topic of active research. When it comes to solid state, the picture becomes even more complicated, as our DFT approach needs to yield accurate description not just for the halogen bond, but for all the other interactions present in the crystal structures (hydrogen bonds, π-π stacking etc.).
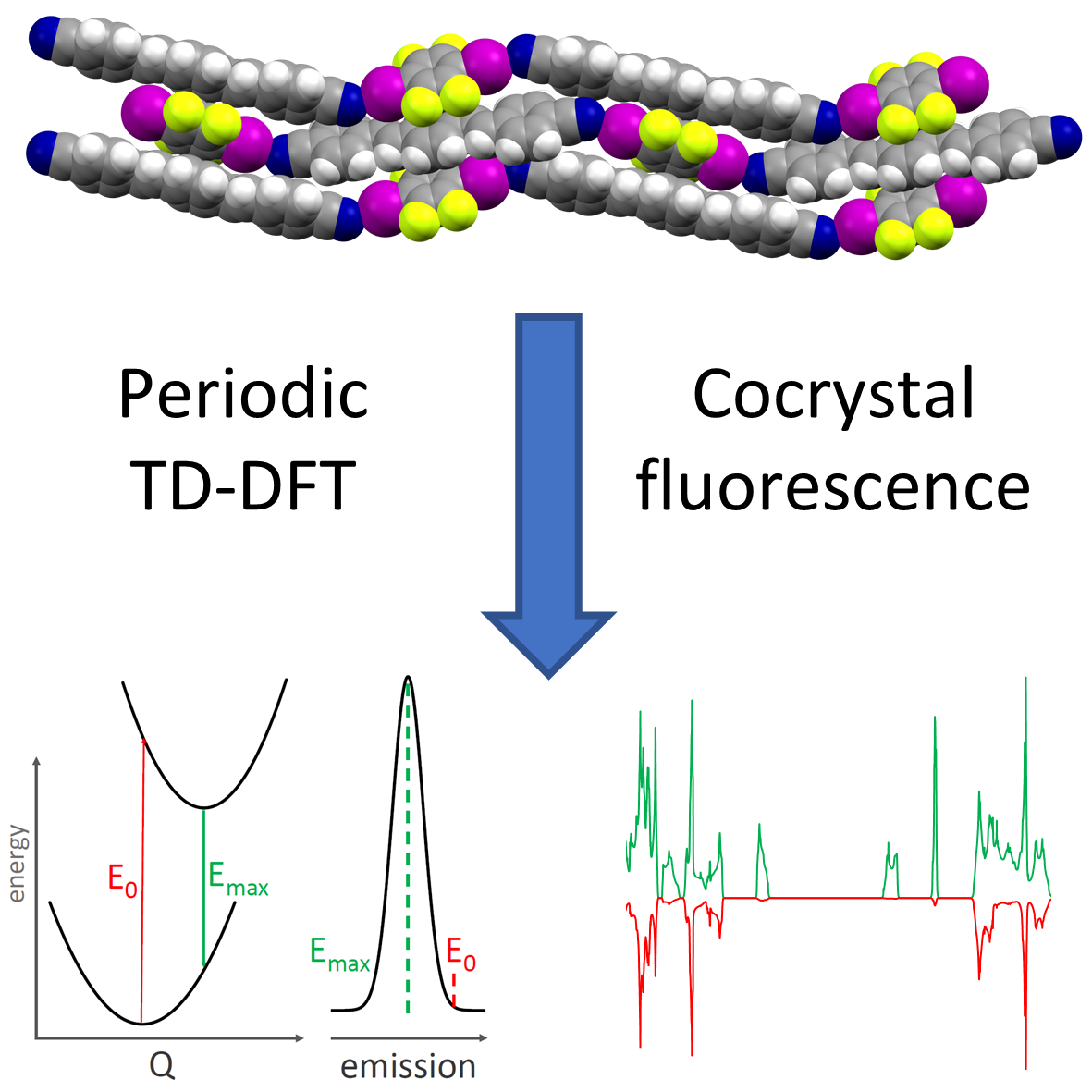
Simulation of solid-state optical and luminescence spectra
Cocrystallization has been shown to yield materials with modified colors and luminescent emission properties, through a modification of crystal packing of chromophore molecules and orbital overlap between cocrystal components. I have shown that periodic time-dependent DFT (TD-DFT) calculations provide for an accurate modelling of solid-state luminescence spectra and explain the relationship between crystal packing and emission wavelength. This methodology allows us to predict solid-state luminescence and design new materials with applications in optoelectronics, display technology and organic lasers.
 https://orcid.org/0000-0003-1150-3108
https://orcid.org/0000-0003-1150-3108